
Abnormalities in brain development, once thought to be irreversible in adults, are assumed to underlie a range of autism, intellectual disability and epilepsy disorders. There is growing evidence that reversing the underlying molecular deficits can result in substantial improvements in function and this knowledge is in turn changing the way we view and envisage treating such disorders in the future.
The aim of the laboratory is to understand the tractability of neurodevelopmental disorders (NDDs) to therapeutic intervention. This includes gaining an understanding of those aspects of NDDs that genuinely result from aberrant early brain development, to features that result from ongoing dysfunction within the mature nervous system. This is important not only for a fundamental understanding of the pathophysiology but also in terms of windows of opportunity for treatment.
To this end, we have worked to develop a gene therapy for Rett syndrome. This is a severe genetic brain disorder that results from disruption of a single gene encoding the epigenetic regulator MECP2. In common with many other developmental disorders, subtle aberrations in transcriptional regulation in neurons can result in dysfunction in neural circuit development and plasticity. Our work seeks to gain a deep understanding of the molecular pathology underlying neurodevelopmental disorders and to then exploit these molecular insights to advance the development of advanced therapies targeting the root-cause of neurological disease.
Specific areas of current investigation within the laboratory include…
Gene therapy for Rett syndrome
Gene therapy for Rett syndrome (RTT), at its simplest, aims to delivery healthy copies of the MECP2 gene to compensate for the mutated ones. Recent developments have shown that achieving widespread brain expression, and maintaining cell-type appropriate control of MeCP2 levels are essential requirements for the successful development of a translational therapy. To achieve this, we are developing expression cassettes capable of producing effective and sub-toxic levels of MeCP2, and combining these with AAV capsids with improved brain penetrance. Our recent studies have shown that a radically truncated MeCP2 is capable of rescuing neurological defects in mouse models of RTT. Minimal MeCP2 may therefore be therapeutically advantageous, and the shorter coding sequence creates room for additional regulatory sequences which may enable more precise control of expression.
Radically truncated MeCP2 rescues Rett syndrome-like neurological defects. (2017) Tillotson R, Selfridge J, Koerner MV, Gadalla KKE, Guy J, De Sousa D, Hector RD, Cobb SR, Bird A. Nature.
Development of a Novel AAV Gene Therapy Cassette with Improved Safety Features and Efficacy in a Mouse Model of Rett Syndrome. (2017) Gadalla KKE, Vudhironarit T, Hector RD, Sinnett S, Bahey NG, Bailey MES, Gray SJ, Cobb SR. Mol Ther Methods Clin Dev.
RNA level therapies
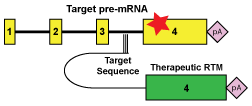
We are developing and assessing the translational potential of alternative therapeutic strategies that target RNA, at the level between the gene and the protein.
Spliceosome-mediated RNA trans-splicing (SMaRT) targets primary RNA transcripts, exploiting natural mechanisms in cells to ‘bypass’ disease-causing mutations. By this method, the gene itself remains under normal regulatory control, ensuring appropriate levels of expression in each cell type. This is important for disorders such as Rett syndrome where the level of the gene product is critical. We have developed tools for the high throughput sceening as well as the optimization of trans-splicing molecules.
Other RNA level approaches being explored in other neurodevelopmental disorders include exon skipping and RNA editing.
mRNA trans-splicing in gene therapy for genetic diseases. (2016) Berger A, Maire S, Gaillard MC, Sahel JA, Hantraye P, Bemelmans AP. Wiley Interdiscip Rev RNA
CDKL5 deficiency
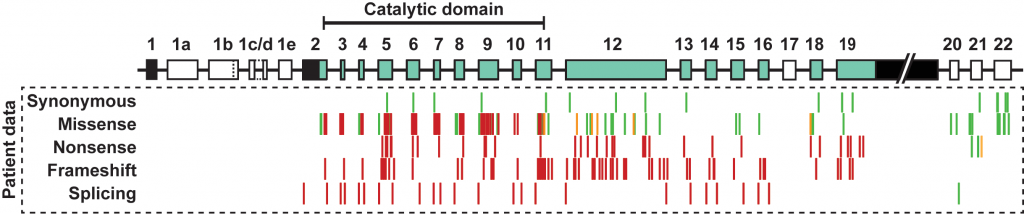
CDKL5 deficiency is a rare, X-linked disorder caused by loss-of-function mutations in the CDKL5 gene. These mutations lead to a characteristic set of symptoms, of which early-onset seizures are a cardinal feature, often presenting as infantile spasms and usually occurring within the first three months of life. Since the identification of mutations in the CDKL5 gene, the disorder has been identified as a distinct clinical entity, and its associated phenotypes have becoming increasingly well-defined over the last decade. We are interested in the genetics of this neurodevelopmental disorder; variants, transcripts and protein isoforms. These aspects of the disorder all inform a rational approach to designing novel genetic therapies targeting CDKL5.
CDKL5 variants: Improving our understanding of a rare neurologic disorder. (2017) Hector RD, Kalscheuer VM, Hennig F, Leonard H, Downs H, Clarke A, Benke TA, Armstrong J, Pineda M, Bailey MES, Cobb SR. Neurology Genetics.
Characterisation of CDKL5 Transcript Isoforms in Human and Mouse. (2016) Hector RD, Dando O, Landsberger N, KIlstrup-Nielsen C, Kind PC, Bailey ME, Cobb SR. PLoS One.
Genetic therapies for other neurodevelopmental disorders
We are developing and evaluating genetic therapies for two further neurodevelopmental disorders. Fragile X syndrome (FXS) is the most common form of inherited intellectual disability and results from genetic silencing in the fragile X mental retardation 1 (FMR1) gene. SynGAP deficiency, caused by mutations in the SYNGAP1 gene, represent a relatively common cause of intellectual disability, epilepsy and autism spectrum disorders. The gene codes for a series of protein isoforms (synaptic Ras GTPase-activating proteins) that have important roles in synaptic function, neurodevelopment and cognitive function. These projects will help us to understand the tractability of FXS and SynGAP1 disorder to genetic intervention at different neurodevelopmental stages and provide a springboard for the generation of novel therapeutic options.